
Bringing emerging nucleic acid technologies to UCI and providing genome-wide analysis for clients interested in gene expression, regulation of gene expression, and genome sequence and variation.
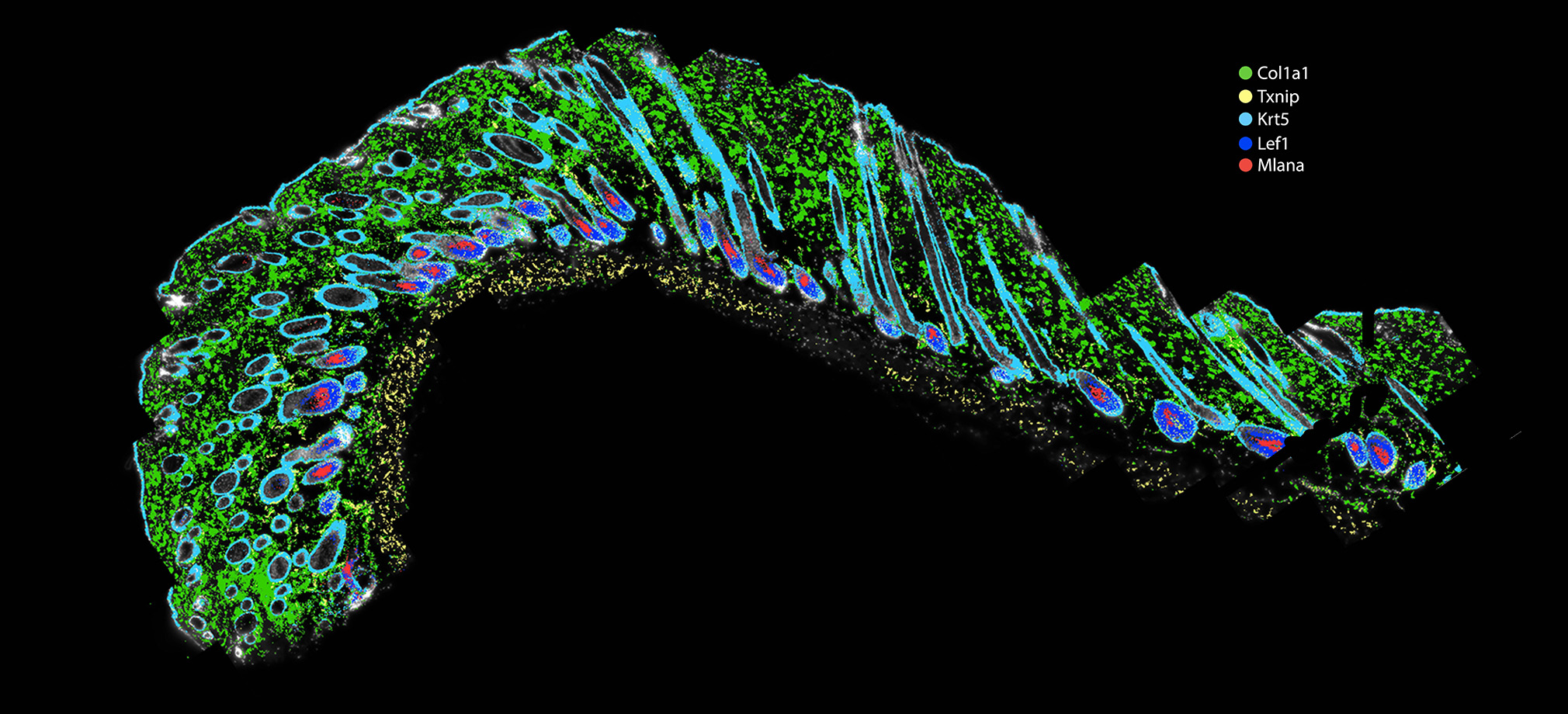


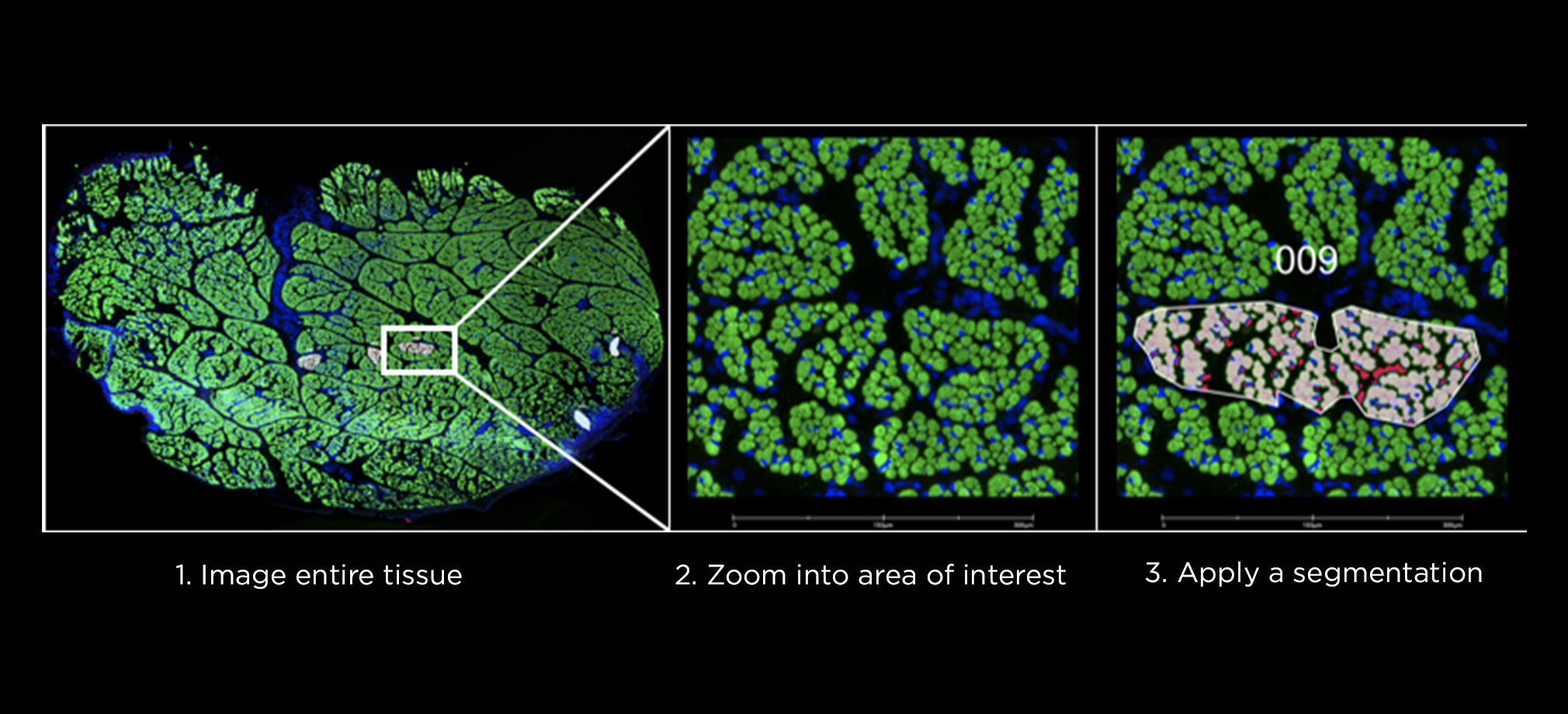

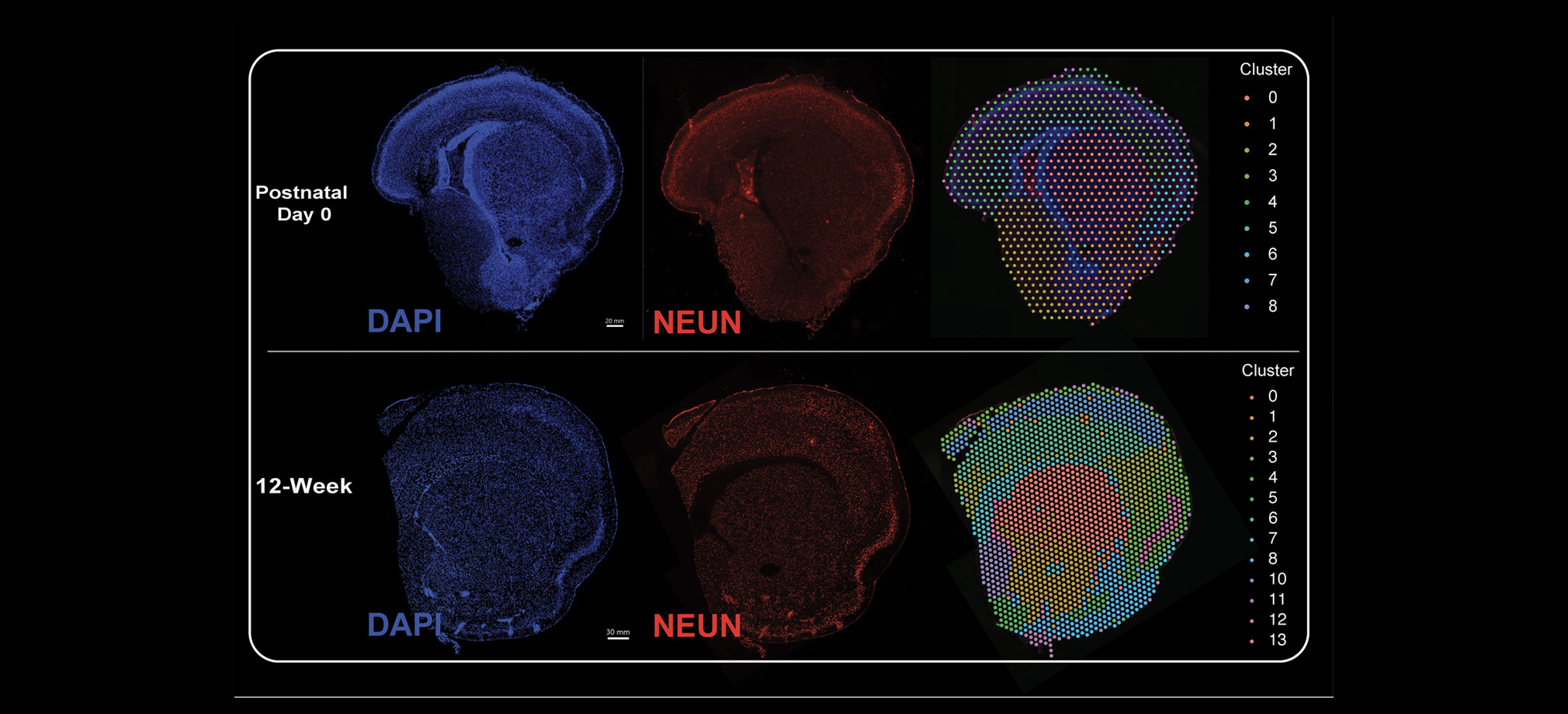
The Genomics Research and Technology Hub (GRT Hub) at the University of California, Irvine is a core research facility. We provide a variety of services ranging from seminar series, workshops, quality checking DNA/RNA, library construction, long and short read sequencing, optical genome mapping, spatial transcriptomics, and bioinformatic analysis. Please explore our site for more information about the instruments and services offered at the GRT Hub. We are available for consultation and look forward to supporting your research projects!
Upcoming Events
Click on an event for more details




Help Us Help You: Acknowledge the Hub in Publications and Seminars
Use iLab to find services, estimate cost and place orders


